A recent study from Ruhr University Bochum sheds light on a novel domain within prion proteins exclusive to mammals, potentially unlocking fresh avenues for investigating neurodegenerative diseases. Prion diseases, characterized by the misfolding of prion proteins, are relentless and ultimately fatal neurodegenerative conditions. While they initially manifest as memory impairments and gait disturbances, they progressively erode essential motor functions and cognitive abilities. Despite their severity, the underlying mechanisms driving their onset remain elusive.
Researchers at Ruhr University Bochum have unveiled a previously unidentified region within prion proteins, offering a promising target for advancing neurodegenerative disease research. This mammal-specific domain influences protein aggregation, a pivotal process implicated in prion disease pathology. Their findings, published on April 22, 2024, in the Journal of Biological Chemistry, mark a significant step forward in understanding these debilitating conditions.
Prion diseases, though rare, exact a devastating toll, affecting approximately 2 individuals per million. Among the most notorious is Creutzfeldt-Jakob disease, thrust into the spotlight during the BSE crisis in the 1990s. As these diseases progress, the brain undergoes a characteristic transformation, exhibiting a sponge-like appearance and accumulating protein deposits.
According to Professor Jörg Tatzelt, who spearheaded the study at the Institute of Biochemistry and Pathobiochemistry, Ruhr University Bochum, “The onset of the disease stems from the misfolding of cellular prion protein, PrP. These aberrant proteins amass in the brain, forming aggregated deposits known as plaques, gradually impairing neuronal function.”
Notably, prion diseases have been exclusively documented in mammals, despite the presence of prion proteins in amphibians and reptiles. Dr. Janine Kamps, the study’s lead author, underscores this evolutionary distinction, stating, “The brains of frogs and reptiles evolved far earlier than those of mammals, prompting us to hypothesize that biophysical and biochemical properties of prion proteins have evolved over time, conferring susceptibility to toxic misfolding solely in mammals.”
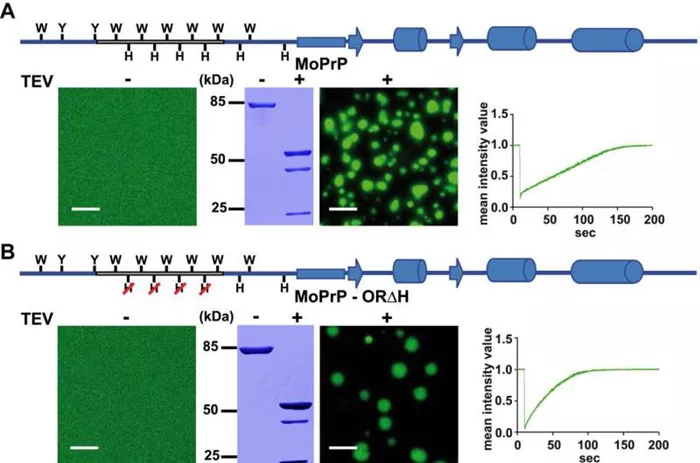
To validate their hypothesis, the researchers conducted fluorescence microscopy analyses of purified prion proteins from frogs and mice, unveiling a distinct protein region unique to mammalian PrP. This region, influencing protein folding and aggregation, signifies a novel function exclusive to mammalian PrP.
“The structure of prions typically exhibits two subdomains—one structured and one intrinsically disordered,” explains Tatzelt. “Of particular interest is the disordered region, the N-terminal domain, which has evolved disparately in mammals compared to amphibians, hinting at a newfound function exclusive to mammalian PrP.”
These findings deepen our understanding of protein folding mechanisms and hold promise for future therapeutic interventions in prion diseases. The researchers’ forthcoming investigations will delve into the precise role of the mammal-specific domain in triggering neurodegenerative diseases, offering renewed hope for mitigating their devastating effects.